前言
我们已经查看了dfHclust 的一些代码。它在很大程度上主要是用于data.frame数据类实例的通用分析:
- 行可以表示要聚类的对象(就是样本名);
- 列可以表示用于比较和分组对象的特征(例如表达值);
rownames可以为对象添加标签;colnames可以给特征值添加名称(例如基因名)。
使用名称这种思路可以很容易在app中方便地查看各种基因的表达值。
当我们对基因组级数据进行分析时就有点不太一样了。我们现在将重点放在表达矩阵方面,它有以下特点:
- 数据通量特别大;
- 假设我们感兴趣的对象是单个生物学样本,它们已经进行了排列(arrayed),那么对它们添加棱可能就比较复杂。原文如下:
assuming the objects of interest are the individual biological samples that have been arrayed, the labeling of objects can be complex
- 假设我们使用ExpressionSet来管理数据,那么
rownamescolnames都将倾向于使用无意义的词汇表来进行标记,例如探针数据标识符(它们都是一些数字)和样本标识符(都是一些表示样本名的英文字符,但无法直接看出是什么样本)。
通常来说,这些都是我们最后遇到的麻烦(除了使用tissuesGeneExpression型数据),这里我们有一个比较简单的解决䢍。我们先来看数据量的问题,我们可以通过使用基于括号的子集(subsetting)来方便地管理ExpressionSet数据。
一个简单的app
现在让我们编写一个函数,这个函数可以将ExpressionSet转换为data.frame,然后使用dfHclust()来处理data.frame。这交进我们的app的一个功能,它用于实验数据中的交互式聚类,如下所示:
|
|
准备组织特异性表达数据
现在我们转换tissuesGeneExpression包中的matrix/data.frame数据,将它们转换为已经注释了的ExpressionSet数据类型,如下所示:
|
|
以下代码是我们随机挑出一些样本的基因集来放到app中运行
|
|
探索组织分化的聚类分析
这个数据集中的哪些基因对于分组来说最为重要?能找到这些基因的分析工具能否帮助我们找到,并理解这些基因呢?这两个问题都不是特别清楚,我们需要理解构成组织基因表达背后的实验设计,并且完全理解它们。然而,以下代码可以帮助我们确定那些在统计学意义不太可能在所有组织中保持恒定表达的基因。我们使用调和F检验(moderated F test)来计算这些基因,它的零假设是,在所有的组织中,基因的表达是恒定的,代码如下所示:
|
|
现在我们根据简单F检验确定了50个最有差异的基因,现在用热图画出来:
|
|
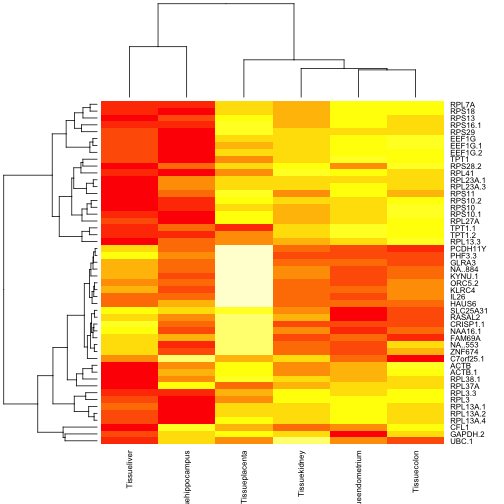
以上是相对于参考分类(小脑,cerebellum)的平均表达值进行的可视化。我们也可以将原始的数据进行可视化,如下所示:
|
|
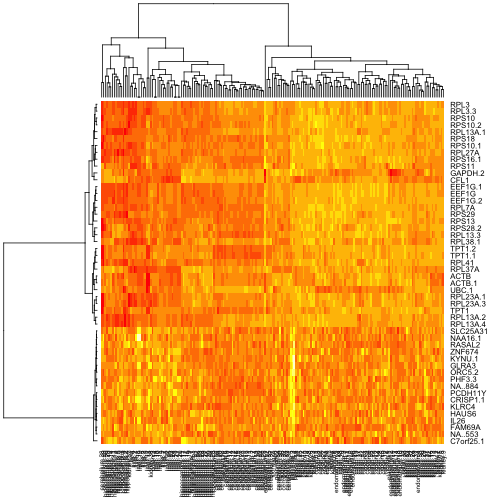
这都是非常非正式的,只使用热图演示的默认值。我们会继续这样做。在检查平均热图时,我认为以下五个基因是组织分化的潜在特征:
这是非正式地展示数据的方法,我们只使用了默认值。 我认为以下五个基因代表了组织分化的潜在特征,如下所示:
原文为:
This is all very informal, using only default values for heatmap presentations. We’ll continue in this vein. In my inspection of the heatmap of means, I considered the following five genes to be a potential signature for tissue differentiation:
|
|
In the exercises we’ll see whether this is at all satisfactory.
机器学习方法评估区分能力
使用机器学习并不是直接解决可视化的问题,但是对于思考我们可以用可视化做哪些事情,它简单且有用。 MLInterfaces 包可以更容易地使用各种基于R的统计学习工具来处理ExpressionSet 实例。我们使用随机森林来检验一下上面定义的sig50 的有效程度。
|
|
这里我们使用了默认值,我们可以看到50个基因标签能够对这几个组织进行了合理的区分。通过使用MLearn对单行代码进行了简单的修改,我们就可以评估这些基因标签改变后以及计算过程的效果。